
Le point sur les amyloses cardiaques
Introduction à destination du cardiologue · Réseau Amylose · Mis à jour déc. 2023
Qu'est-ce que l'amylose cardiaque ?
L’amylose est la manifestation de plusieurs maladies systémiques (article Mohty D et al ACVD).
La caractéristique commune de ces maladies est une accumulation extracellulaire de protéines fibrillaires insolubles qui se déposent et envahissent progressivement les tissus empêchant leur bon fonctionnement. L’infiltration amyloïde cardiaque est responsable d’un épaississement du myocarde créant une « hypertrophie ».
Cette infiltration se complique d’insuffisance cardiaque et de trouble de la conduction. Tout ceci explique pourquoi le cardiologue est en première position pour faire le diagnostic de cardiopathie amyloïde. Malheureusement ce diagnostic est souvent évoqué tardivement après un diagnostic initial « d’insuffisance cardiaque à Fraction d’Éjection Ventriculaire Gauche (FEVG) préservée » ou de « cardiopathie hypertensive ».
Afin d’améliorer l’orientation diagnostique devant une cardiomyopathie et de dépasser ces simples diagnostics, la Société Européenne de Cardiologie (ESC) a récemment revu la définition et la classification des cardiomyopathies.
Les cardiomyopathies hypertrophiques (CMH) sont définies désormais par un simple épaississement du myocarde regroupant ainsi sous le terme CMH des processus physiopathologiques très différents allant d’une hypertrophie des cardiomyocytes observée dans les CMH sarcomériques (mutation troponine, myosine…) à l’infiltration du myocarde.
Points clés pour les professionnels de santé
L’amylose cardiaque est sous-diagnostiquée : elle se cache derrière des atteintes ORL (surdité), rhumatologiques (canal carpien, doigt à ressaut, Dupuytren, prothèse, …), des tableaux d’insuffisance cardiaque à FEVG préservée, de cardiomyopathie hypertrophique ou de rétrécissement aortique à bas débit.
Les deux formes principales sont l’amylose AL (60 % des cardiopathies amyloïdes) et l’amylose ATTR (sauvage ou héréditaire). Leur diagnostic différentiel est indispensable. Les traitements sont radicalement différents.
Des traitements spécifiques et efficaces existent. La précocité du diagnostic conditionne le pronostic. La prise en charge cardiologique de l’amylose diffère de celle de l’insuffisance cardiaque classique.
Quand penser à l'amylose cardiaque ?
La Société Européenne de Cardiologie (ESC) a revu la classification des cardiomyopathies. Les CMH sont désormais définies par un épaississement du myocarde, regroupant des mécanismes très différents — de l’hypertrophie des cardiomyocytes à l’infiltration.
Évoquer une amylose cardiaque devant :
Toute CMH (cardiomyopathie hypertrophique)
Insuffisance cardiaque à FEVG préservée
Insuffisance cardiaque à FEVG altérée avec HVG
Toute hypertrophie myocardique symptomatique
Hypertrophie Ventriculaire Gauche HVG + BSA3 ou BAV2-3 ou FA ou flutter
Rétrécissement aortique + HVG ou bas débit / bas gradient
Pourquoi dépister l'amylose cardiaque ?

Classification des amyloses — 4 types principaux
La classification des amyloses repose sur la nature biochimique de la protéine amyloïde impliquée dans la formation des dépôts. Une vingtaine de protéines peuvent former des fibrilles amyloïdes (Fibrinogène, apo A1…). Les formes cardiaques les plus fréquentes sont les amyloses AL (immunoglobuliniques) et les amyloses à transthyrétine (TTR) : héréditaire (TTR mutée) ou sauvage (TTR sauvage).
Type d’amylose | Amylose AL | Amylose à transthyrétine | Amylose AA | |
Héréditaire | Sauvage | |||
Dépots | Chaînes légères d’Ig(Kappa ou Lambda) | Transthyrétine mutée | Transthyrétine sauvage | Protéine inflammatoire |
Source | Moelle osseuse | Foie | Foie | Protéine inflammatoire |
Organes atteints | Coeur, rein, foie, système nerveux | Système nerveux périphérique, coeur | Coeur | Rein, Foie |
Sources, type de dépôts et organes atteintes dans les quatres types principaux d’amylose
Les amyloses AL
Les amyloses AL sont liées principalement aux gammapathies monoclonales (MGUS) ou au myélome. Les gammapathies monoclonales sont très fréquentes et touchent environ 10% des patients de 60 ans. Ces gammapathies se compliquent heureusement rarement d’amylose mais représentent plus de 60% des cardiopathies amyloïdes.
Les amyloses ATTR sauvage (ATTRwt)
La transthyrétine (TTR) est une protéine synthétisée par le foie sous forme de monomère. Ces monomères s’assemblent en tétramères qui transportent des protéines (ex : hormone thyroïdienne, vitamine D) dans le sang. Les amyloses TTR sont de deux types :
1/ Amylose à transthyrétine dite « sénile » ou « sauvage » où le précurseur est la transthyrétine non-mutée (ATTRwt) survenant quasi-exclusivement chez des hommes âgés. (article Damy T et al, Senile systemic amyloidosis). Les mécanismes de cette amylose ne sont pas connus.
2/ Amylose à transthyrétine héréditaire, la forme familiale où la transthyrétine est mutée (ATTRm) (article Mohty D et al, cardiac amyloidosis).
La transmission est autosomique dominante. Plus de 120 mutations pathogènes du gène codant pour la TTR ont été identifiées. Les dépôts tissulaires comportent également de la transthyrétine non mutée. Bien qu’héréditaire, on ne retrouve des antécédents familiaux que dans 50% des cas. L’atteinte tissulaire est variable suivant la mutation.
La mutation ATTR Val122Ile est la plus fréquente. Cette mutation est présente chez 3,9% des patients d’origine africaine (article Jacobson DR et al, variant-sequence transthyretinin late-onset cardiac amyloidosis in black Americans). La pénétrance de ces mutations est variable suivant le fond génétique et la mutation. Les cas sporadiques se révèlent généralement tardivement, c.à.d. après 50-60 ans.
La mutation ATTR Val30Met est fréquente chez les patients d’origine portugaise par une neuropathie à l’âge de 25 à 30 ans. Certaines mutations (Val122Ile, Ser77Tyr…) touchent principalement ou exclusivement le cœur. Ces cas d’amylose sont sous-estimés dans la population des patients cardiologiques.
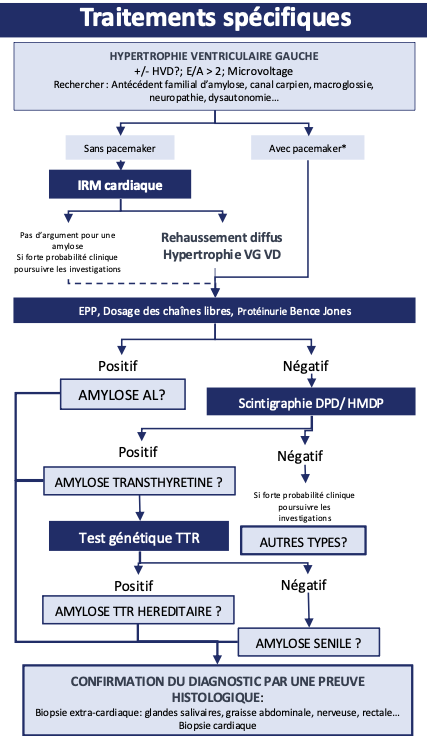
Le diagnostic se fait sur l’analyse anatomopathologique qui retrouve des dépôts d’amylose avec un immunomarquage positif pour transthyrétine. Le test génétique réalisé par séquençage qui identifie la mutation du gène de la transthyrétine.
Orientation diagnostique
La recherche systématique des signes cliniques, ECG et échocardiographiques devant toute hypertrophie ventriculaire permet un diagnostic plus précoce.
Pourquoi un centre de référence ?
Prise en charge multidisciplinaire
Cardiologie, hématologie, neurologie, génétique, anatomopathologie — sur un même site
Accès rapide aux soins
Cardiologie, hématologie, neurologie, génétique, anatomopathologie — sur un même site
FAQ
Devant toute CMH inexpliquée, insuffisance cardiaque à FEVG préservée, HVG avec troubles de conduction (BSA, BAV, FA), ou rétrécissement aortique à bas débit et bas gradient. Souvent diagnostiquée tardivement après un diagnostic d’insuffisance cardiaque ou cardiopathie hypertensive.
La forme sauvage (ATTRwt) est liée à un dépôt de TTR non mutée chez l’homme âgé, mécanismes non élucidés. La forme héréditaire (ATTRv) est liée à une mutation du gène TTR (>120 mutations, autosomique dominante) avec atteinte neurologique et/ou cardiaque selon la mutation.